
Les accidents de l’ère industrielle de la pharmacie posent la question de la mise en place d’une sécurité systémique dans ce domaine telle qu’elle existe dans les domaines du nucléaire civil, du spatial et de l’aérospatiale. La pharmacovigilance a pour objet la surveillance des médicaments et la prévention du risque d’effet indésirable résultant de leur utilisation, que ce risque soit potentiel ou avéré. Elle constitue une garantie qui s’exerce tout au long de la vie d’un médicament. Elle s’inscrit donc dans la science relative à la détection, l'évaluation, la compréhension et la prévention des effets indésirables ou tout autre problème lié aux médicaments.
Par lettre du 26 janvier 2013, la Ministre de la Santé française a confié au Directeur Général de la Santé (DGS) une mission relative à la refonte des vigilances sanitaires. Cette mission a pour objectif d’identifier les moyens permettant de : faire des patients des acteurs de la politique de santé et de la sécurité sanitaire, en facilitant les signalements qu’ils effectuent ; promouvoir l’implication des professionnels, quel que soit leur mode d’exercice, dans le signalement des événements indésirables ; préciser le rôle des agences régionales de santé (ARS : Agences Régionales de Santé) tant pour le partage des signalements que pour la gestion des signaux et alertes ; optimiser le système d’information dans le sens d’une meilleure exhaustivité, d’une plus grande pertinence des signaux recueillis et de leur traitement, en formalisant les conditions de recours aux niveaux national et régional ; réorganiser la chaîne de traitement des signaux par les différentes agences nationales, au premier rang desquelles l’ANSM et l’InVS, en précisant l’articulation des compétences respectives ; clarifier les financements.
Les essais cliniques font partie des domaines spécifiques de management du risque. La vision transposée de Rasmussen (cf. Figure 3), replacée dans la vue générale de la pharmacovigilance, s’attache à souligner l’importance de la transmission d’informations au sein du système socio-technique, particulièrement dans la phase de développement du système, en l’occurrence dans la phase de développement clinique des produits de santé.

La pharmacovigilance est la surveillance des médicaments et la prévention du risque d’effet indésirable résultant de leur utilisation, que ce risque soit potentiel ou avéré.
Elle repose sur :
– le recueil des effets indésirables basé sur la notification spontanée par les professionnels de santé, les patients et associations agréées de patients et les industriels avec l’appui du réseau des 31 centres régionaux de pharmacovigilance,
– l’enregistrement et l'évaluation de ces informations, la mise en place d’enquêtes ou d’études pour analyser les risques, la participation `a la mise en place et au suivi des plans de gestion des risques,
– l’appréciation du profil de sécurité d’emploi du médicament en fonction des données recueillies,
– la prise de mesures correctives (précautions ou restriction d’emploi, contre-indications, voire retrait du produit) et la communication vers les professionnels de santé et le public,
– la communication et la diffusion de toute information relative à la sécurité d’emploi du médicament,
– la participation à la politique de santé publique de lutte contre la iatrogénie médicamenteuse (la iatrogénie médicamenteuse désigne les effets indésirables provoqués par les médicaments).
Les SARs, les réactions sérieuses, attendues qui ne sont ni fatales ni mortelles doivent déclarées dès que possible, mais pas au-delà de 15 jours civils (calendaires) après la première connaissance par le promoteur. En revanche les SUSARs fatals ou mortels, inattendus arrivant dans essais cliniques doivent être notifiés (par e-mail, téléphone, télécopie, ou par écrit) d`es que possible, mais pas au-delà de 7 jours civils après la première connaissance par le promoteur. Un rapport complémentaire aussi complet que possible peut suivre dans 8 jours calendaires supplémentaires. Le promoteur déclare, le fait, à l’autorité grâce au formulaire CIOMS-I (Suspect Adverse Reaction Reaction). Le moniteur devra s’assurer sur site que ces documents ont été réceptionnés et lus par l’investigateur. Ces notifications sont `a classer dans l’ISF (Investigator Site File).
Au-delà du projet concerné par l’essai clinique courant, il existe une communauté regroupant tous les acteurs de tous les protocoles qui sont conduits avec ce produit.
Une copie du formulaire CIOMS-I associé à tout SUSAR (Suspected Unexpected Serious Adverse Reaction) survenant dans l’essai clinique courant sera envoyée électroniquement par le promoteur `à cette communauté :
– à l’état membre dans lequel la réaction est arrivée,
– aux autres états membres concernés,
– à l’Agence (Eudravigilance CT Module),
– aux investigateurs et moniteurs utilisant le produit (dans les autres protocoles).
Le formulaire CIOMS-I comporte des informations codifiées en particulier la catégorisation de l’événement indésirable à partir du Verbatim du médecin investigateur.
Ce codage des évènements indésirables répond à un standard édicté par les Bonnes Pratiques Cliniqus ICH. Il requiert l'utilisation d'un dictionnaire électronique disponible sous forme d'un logiciel certifié, le MedDRA.

Le MedDRA ou Dictionnaire Médical des Affaires Réglementaires (en anglais Medical Dictionary for Regulatory Activities) est une terminologie médicale standardisée développée par le Conseil international d'harmonisation des exigences techniques pour l'enregistrement des médicaments à usage humain (CIH -en anglais ICH pour International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use), afin de faciliter au niveau international le partage d’informations réglementaires concernant les produits médicaux à usage humain. Cet outil est utilisé par les autorités réglementaires et l'industrie pharmaceutique pendant le processus réglementaire, depuis les études pré-commercialisation jusqu'aux activités post-commercialisation, en facilitant la saisie des données, ainsi que leur recherche, leur évaluation et leur présentation. En outre, il s'agit du dictionnaire approuvé par le CIH pour la classification des évènements indésirables.
Initialement disponible en anglais et en japonais, le MedDRA est maintenant traduit en chinois, en tchèque, en néerlandais, en français, en allemand, en hongrois, en italien, en portugais et en espagnol. Le MedDRA est très largement utilisé à l'échelle internationale, y compris aux États-Unis, dans l'Union Européenne et au Japon. Son utilisation est actuellement obligatoire en Europe et au Japon pour les déclarations de pharmacovigilance.
Le dictionnaire MedDRA est organisé par disciplines médicales (SOC pour System Organ Class), divisées en groupes de termes de haut niveau (HLGT pour High-Level Group Terms), en termes de haut niveau (HLT pour High-Level Terms), en termes préférentiels (PT pour Preferred Terms) et finalement en termes de plus bas niveau (LLT pour Lowest Level Terms)2. De plus, le dictionnaire MedDRA contient des requêtes MedDRA standardisées (SMQ pour Standardized MedDRA Queries). Ces SMQs sont des groupements de termes qui se rapportent à une condition médicale ou à un centre d'intérêt définis.
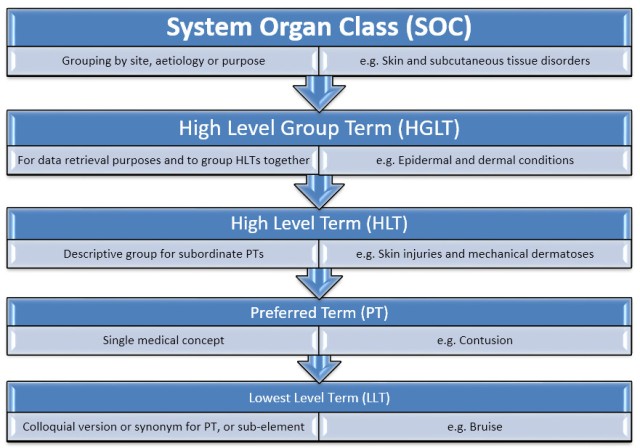
Les cas sont habituellement codés lors de la saisie des données au niveau le plus spécifique (LLT), et les évènements ou cas sont en général comptabilisés au niveau des PT. Les plus haut niveaux (HLT, HLGT et SOC) ainsi que les SMQs sont utilisées pour faire des recherches dans les bases de donnée, organiser les résultats et calculer des sous-totaux.
Le codage par une tierce partie indépendante est un gage de confiance et de neutralité dans le codage des évènements. Ce codage peut avoir des conséquences sur le traitement de l'information critique de sécurité et donc sur la sécurité du produit santé.
Pour cette raison nous préconisons une indépendance de partie dans le traitement et le codage de ces informations de sécurité sanitaire. Un tiers de confiance, une block chain à même de ré-assurer les parties prenantes du promoteur pharmaceutique.
Est-il possible de créer du sens par un processus d’intelligence collective qui dépasse les murs de l’entreprise productrice des produits de santé et ainsi faire émerger un processus de veille stratégique (Amabile, 1999) au niveau des parties prenantes (Laboratoires promoteurs, CRO Contrat Research Organisation et autorités de santé) ? Ce processus, (Arena et al., 2015), serait il apte à prévenir des évènements indésirables graves affectant des sujets-patients ? Mais aussi à détecter, prévenir ou attester de fraudes ou mauvaises conduites dans des essais inéthiques ; et ainsi maintenir la démarche de recherche clinique dans une « enveloppe de sécurité acceptable » ?
Tel est le défi sécuritaire des industries et du système de santé à l’horizon 2030 auquel nous
souhaitons prendre part.
#
Un contenu,
une proposition,
un service de

Tous droits réservées.











